


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
藤野 威男; 田川 博章
Analytica Chimica Acta, 107, p.365 - 371, 1979/00
被引用回数:3アルカリ土類金属を使った酸化重量法の適用性を3元系ウラン酸化物M-U-O(M=La,Ce,Th)について調べた。方法はMgOあるいはBa

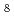 UO
UO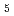 を酸化物試料と混合し、空気中適当な条件下で加熱する。アルカリ土類金属の共存下において加熱すれば、反応によってウランは完全に6価まで酸化されるから、反応前後の重量を測定すれば試料の酸素量が求められる。La
を酸化物試料と混合し、空気中適当な条件下で加熱する。アルカリ土類金属の共存下において加熱すれば、反応によってウランは完全に6価まで酸化されるから、反応前後の重量を測定すれば試料の酸素量が求められる。La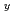 U
U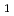
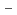 O
O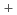
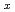 の酸素はMgOの添加によりy=0.8までx値の標準偏差
の酸素はMgOの添加によりy=0.8までx値の標準偏差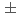 0.006で定量された。ThyUOの酸素はBaUOの添加によりy=0.8までx値の標準偏差0.01で定量された。CeO-UO系についても分析を行った。
0.006で定量された。ThyUOの酸素はBaUOの添加によりy=0.8までx値の標準偏差0.01で定量された。CeO-UO系についても分析を行った。
町田 昌彦; 中村 博樹
no journal, ,
Thorium has been considered as an alternative nuclear fuel element to uranium. Thorium is well-known to be more abundant in nature than uranium. Furthermore, it is widely spread that thorium dioxide is chemically more stable than uranium dioxide. From an economical and secure point of view, thorium fuel can be a candidate fuel in next-generation nuclear reactors. However, the data of thermal properties of thorium dioxide are insufficient, in particular, in the high-temperature region near its melting point. In order to supplement such thermal data of the materials, classical molecular dynamics have played an important role and been applied also to thorium compounds. However, the thermal properties obtained by classical molecular dynamics often depend on the empirical parameters giving the atomic potential. Especially, it is known that the Bredig transition (rapid growth of heat capacity slightly below melting point) strongly depends on the choice of parameters of the atomic potential in the case of molecular dynamics of uranium dioxide. Thus, classical molecular dynamics is not so reliable for high-temperature behaviours, such as the Bredig transition. Therefore, we adopt first-principles molecular dynamics to avoid this parameter sensitivity. In the present paper, we evaluate thermal properties of thorium dioxide at high temperature using first-principles molecular dynamics. The calculated enthalpy agrees well with the observed data. We find that the obtained Bredig transition temperature coincides with the experimental values and that this transition is caused by the high mobility of oxygen by visualizing the oxygen motions. Consequently, we reveal that the first-principles molecular dynamics can provide reliable data of thermal properties of nuclear fuels even at high temperature.
小林 恵太*; 奥村 雅彦; 中村 博樹; 板倉 充洋; 町田 昌彦
no journal, ,
二酸化トリウムは高い融点、化学的安定性、またウランに比べ埋蔵量が豊富であることから、二酸化ウランに代わる核燃料の一つの候補であると考えられている。しかし、高温下でのトリウム熱物性に関するデーターは十分に得られているとは言えない。高温下での物性データーは簡単には取得できないため、分子動力学による原子レベルでのシミュレーションが有用となるが、通常の古典分子動力学は経験的なパラメーターに強く依存するため、得られた結果の信頼性には十分な検討が必要となることが問題となっていた。本発表では、第一原理計算と同等の精度で分子動力学の実行が可能となる、機械学習分子動力学法による二酸化トリウムの高温物性の解析を行った。その結果、得られた物性値は実験値とよく一致した。本研究の結果は、機械学習分子動力学法によるシミュレーションが熱力学的な物性値の評価に用いることを示唆しており、今後、経験的なパラメーターによらない燃料物性評価シミュレーションの可能性が拓かれたと言える。
